Influenza virus – how epidemics and pandemics emerge
Influenza virus has been described as the last great plague. It is characterised by an ability to constantly change its two surface proteins – haemagglutinin and neuraminidase – allowing the virus to cause successive epidemics every one or two years or more serious pandemics at irregular intervals. This phenomenon of successive infections by the influenza virus is in marked contrast to the situation with viruses like measles, mumps or small pox where exposure to a single infection induces lifelong immunity.
In the period from 1975 to 1983, CSIRO scientists Colin Ward, Theo Dopheide, Gerry Both and Merilyn Sleigh achieved international recognition for their work on the amino acid and gene sequences of numerous influenza virus haemagglutinins. Their data showed conclusively that the emergence of new influenza virus epidemics was associated with the accumulation of point mutations in the virus coat proteins. In contrast the emergence of major pandemics was the result of a different mechanism, genetic re-assortment. The sequence data was used also by Don Wiley (Harvard University) and colleagues in their landmark determination of the 3D structure of the influenza virus haemagglutinin, published in Nature in 1981.
Influenza viruses
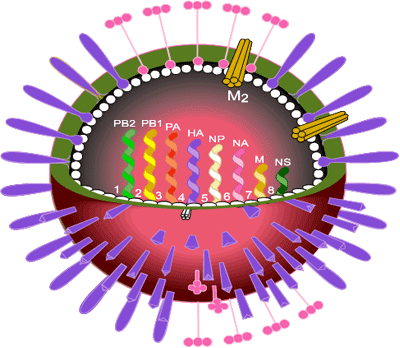
The influenza virus occurs as three types A, B and C which correspond to three genera of the family Orthomyxoviridae. Type A is the cause of epidemics and pandemics and infects animals and birds as well as humans. Type B can cause epidemics but does not infect other animals or birds. Type C can cause mild infections in humans but does not cause epidemics. The genomes of all influenza viruses consist of single-stranded, negative sense RNA. In influenza A viruses there are 8 segments of RNA coding for eight viral proteins and two non-structural proteins.
Influenza virus is a pleomorphic, enveloped virus with two coat proteins on its surface, the haemagglutinin (HA) and the neuraminidase (NA). HA is the major coat protein (~95% of the total) and is responsible for cell attachment and infection by a membrane fusion process. It is the main target of protective immune reactions and thus was the main focus of attention at that time.
Virus strains are named according to influenza virus type, the place where first isolated, the isolate number and the year of isolation as well as the nature of the two surface proteins. Examples are the Hong Kong virus strain A/England/102/72 (H3N2). The Asian influenza viruses which circulated in man from 1957 to 1968 were H2N2 and the viruses preceding Asian influenza (including the lethal Spanish influenza of 1918) were H1N1, as was the swine influenza pandemic of 2009.

Early controversy
In the early 1970s there was considerable speculation about the way in which the influenza virus escaped immune responses and continued to cause repeated infections year after year. The reason for this is not a poor immune response, rather it is the fact that the influenza virus continues to change its coat proteins so that the new infecting variants are no longer recognised and destroyed by the immune response generated against the earlier infection.
In a series of stimulating papers, Dr Stephen Fazekas de St Groth, at the CSIRO Division of Animal Genetics, had proposed a single mechanism (accumulated point mutations) for the two types of antigenic variation seen in influenza virus: antigenic drift which occurs within influenza virus subtypes and antigenic shift to new subtypes such as the emergence of Asian influenza in 1957 and Hong Kong influenza in 1968. This theory was based on the asymmetric nature of the antigenic cross reactions observed between parent viruses and their mutants selected in the presence of neutralising antibodies. This work was widely publicised in the popular press. Fazekas believed that new epidemic strains could be generated in advance and this led to the Pasteur Institute manufacturing a vaccine from one of Fazekas’s ‘prospective’ strains.
At the same time Dr Graeme Laver at the John Curtin School of Medical Research, Australian National University, and Dr Robert Webster from St Jude Children’s Research Hospital, Memphis, Tennessee had obtained evidence (using peptide mapping) for a different mechanism for antigenic shift. The mechanism they proposed was genetic re-assortment and a major controversy developed that required resolution.
Since influenza A viruses contain eight separate segments of RNA, genetic re-assortment can occur when cells are infected simultaneously by two or more influenza viruses resulting in progeny viruses that contain some (1, 2, 3 or 4) RNA segments from one parent virus and the remaining (7, 6, 5 or 4) RNA segments from the second virus. This phenomenon had been demonstrated experimentally years before by Macfarlane Burnet at the Walter and Eliza Hall Institute in Melbourne.
The project begins
In the period 1972-83 CSIRO was centrally involved in the international research effort that solved the controversy and established the molecular mechanisms behind both antigenic shift and drift. The program involved protein chemical studies by Colin Ward and Theo Dopheide at the Division of Protein Chemistry in Parkville (1972-81) and nucleic acid cloning and sequencing by Gerry Both and Merilyn Sleigh at the Division of Molecular Biology at North Ryde (1978-83).
The suggestion to initiate the protein work came from the Chief of Protein Chemistry, Gordon Lennox, in 1972 after discussions with several Chiefs of CSIRO about possible projects where protein chemical investigations might be of value. Colin Ward visited Stephen Fazekas late in 1972 to discuss Fazekas’s theory and a possible collaboration. As Colin recalled:
Stephen was from a Hungarian aristocrat family and was an imposing figure. His lab was in a small building half-way down the hill at North Ryde just above the poultry sheds. When I entered his lab he was smoking a cigar with the music of Bella Bartok playing in the background. Within minutes he had mentioned that there was this fellow in Canberra, Graeme Laver who had different views and that I should not take too much notice of what he said. About three weeks later I received a phone call from Graeme Laver commenting that he had heard I was going to sequence the haemagglutinin. He quickly went on to add that I should not take too much notice of what Stephen Fazekas said. This was wonderful, here was a raging controversy involving an important human health problem that was crying out for experimental data.
In 1973, Colin spent a week in Graeme Laver’s laboratory at the John Curtin School of Medical Research, Australian National University to learn how to culture and purify the virus and its proteins. During the previous decade Laver had established the methods for:
- purification of the virus by affinity binding to chicken red blood cells and virus elution from the resuspended red blood cells by incubating at 37 °C allowing the viruses own neuraminidase to remove the terminal sugar residues to which the HA was bound
- purification of HA by electrophoresis on thin cellulose acetate sheets
- isolation of the heavy (HA1) and light (HA2) chains of influenza virus HA by overnight density gradient centrifugation in 6M guanidine hydrochloride after mild reduction.
The method for growing the virus (48 hours culture in inoculated 10-day chick embryos), was the same as that used by the vaccine companies today and was worked out by Macfarlane Burnett while at the National Institute of Medical Research, London from 1932 to 1933.
The size of the challenge
At the time the work commenced it was known that the HA monomer was a two chain protein containing a heavy chain (HA1) and a light chain (HA2) held together by disulphide bonds. Size estimates at the time suggested HA1 contained over 500 amino acids and HA2 approximately 300 amino acids making its sequence determination by Edman chemistry a formidable challenge.
As Colin recalled, he had two choices – develop more sensitive methods to allow protein sequencing on a smaller scale; or – get lots of protein. He chose the latter and during the subsequent nine years used over 150 000 chick embryos to grow the 10 grams of virus (1 gram of HA) required. He had more influenza virus in his fridge than anyone else in the world bar the vaccine companies.
In 1975 Colin was joined by Theo Dopheide, an experienced protein chemist, who had been working on sequences of the matrix proteins of the wool fibre. This turned out to be a very productive partnership as they complemented each others skills effectively. Colin was a biologist and biochemist while Theo’s strengths were chemistry and an ability to maintain electronic instrumentation.
A major challenge was to scale-up the isolation of HA from the virus particles. Laver’s cellulose acetate electrophoresis in the detergent SDS was the method of choice, but the sheets were so thin that their use would be impractical – it would require thousands and thousands of runs. The solution came in the form of 5 mm thick cellulose acetate blocks pre-equilibrated in buffer solution which allowed tens of milligrams of solubilised virus to be processed daily. The isolated HA was then separated into the two chains by Laver’s method of overnight high-speed centrifugation in guanidine hydrochloride density gradients. Almost two years were spent generating the protein required for the sequencing effort and it was not without drama. As Colin recalled:
We initially worked on the heavy chain, on the five fragments produced by cyanogen bromide cleavage. When Theo completed the three C-terminal fragments (which accounted for 68 amino acids) he decided to start on the light chain which had been stored in 6M guanidine hydrochloride in a small flask in a fridge at the end of the second floor of the Parkville laboratory. I can still see it vividly – as he opened the fridge door the flask, which over time had moved to the edge of the top shelf, fell to the floor and shattered. There in the dust and grime was the HA light chain – the result of almost 2 years of sustained effort of virus production and protein purification. I felt sick. But Theo without dropping a beat said “Come on mate, we can scrape it up”. And he did – ending up with as much of the now brown, discoloured solution as possible. The pure protein was recovered following overnight precipitation with ethanol at minus 20 ºC and we were back in business.
The international influenza meetings of 1977 and 1979
The CSIRO sequencing studies focused on strains of the Hong Kong H3 subtype which appeared in man in 1968 replacing Asian influenza (H2N2) which swept the world in 1957. New epidemics of Hong Kong influenza have appeared every couple of years since.
The H3 haemagglutinin sequence data was first presented by Colin Ward at a specialist symposium at Baden near Vienna in March 1977. On route to this meeting he visited Mike Waterfield at the Imperial Cancer Research Fund, in London who was sequencing the HA protein from the Asian influenza virus A/Japan/57 (H2N2) and for the first time was able to see the relationship between these two proteins – the sequence identities were approximately 50%. In December 1979 a second International influenza meeting was held at Thredbo, Australia. Both the Vienna and Thredbo meetings were attended by most of the world’s leading influenza virus researchers. As Colin recalled:
We were fortunate to be collaborating with Graeme Laver who organised both of these events. Theo and I were newcomers to the field and at the time of the Vienna meeting had only published one paper – not enough to get an invitation. But because of Graeme, I was able to be there from the beginning and hear about the exciting things that were happening with the influenza virus – ranging from the selection of variants in response to antibody pressure through to protein crystallography.
By the time of the Thredbo meeting there was a lot of sequence data available and Graeme organised for a large supply of amino acid 3-letter code cards to be printed so that the sequences could be put up on the walls of the hall. They were and remained there for the duration of the conference.

The emergence of epidemic strains (antigenic drift) involves accumulated point mutations
Complete amino acid sequences were obtained for the HAs of the early Hong Kong influenza virus A/Aichi/68 and a subsequent major epidemic strain, A/Memphis/72. The Memphis/72 sequence was done first and took Colin and Theo six years to complete. The A/Aichi/68 sequence took just six months because they could make use of the framework provided by the Memphis/72 data. The third complete sequence determined, that of A/duck/Ukraine/63, took only six weeks.
Colin and Theo were able to interpret the peptide maps generated by Drs Gillian Air and Graeme Laver (Australian National University) in collaboration with Dr Robert Webster (St Judes Childrens Hospital, Memphis, Tennessee) and Dr Walter Gerhard (The Wistar Institute, Philadelphia, Pennsylvania). This quantitative peptide mapping data revealed the partial amino acid sequences of nine Hong Kong subtype field strains isolated between 1968 and 1977 and ten laboratory mutants selected by culturing in the presence of monoclonal antibodies. In the monoclonal antibody work Laver, Air, Webster and Gerhard redirected their research program from the H1 sub-type to the Hong Kong H3 sub-type because of the availability of the sequence data being generated by CSIRO on the H3 viruses.
These studies were extended by Gerry Both and Merilyn Sleigh who analysed the sequences of the complete HA gene from five Hong Kong (H3) viruses and of the gene segment coding for the heavy chain of a further 14 Hong Kong influenza strains selected between 1968 and 1980.

These data on natural variants and laboratory mutants selected under antibody pressure revealed that antigenic drift involved the progressive accumulation of point mutations at multiple positions in the HA protein sequence. Each new epidemic strain differed by about 1-2% from the previous strain. Successive strains were not generated by further mutations at the same position as envisaged by Stephen Fazekas, rather they retained the mutations of their predecessor and accumulated new mutations at new positions in the protein sequence.
The emergence of new pandemic strains (antigenic shift) involves genetic re-assortment
Comparison of the CSIRO H3 sequences with those of the Asian influenza virus A/Japan/57 (H2N2) being determined by Mike Waterfield in London established that the mechanism behind the emergence of new pandemics, antigenic shift, was different. The H2 and H3 protein sequences differed by more than 50% supporting the Laver-Webster theory of genetic re-assortment. Using peptide maps Laver and Webster had suggested that the H3 gene of the 1968 human Hong Kong pandemic virus may have come from the bird virus (A/duck/Ukraine/1/63) isolated five years before the appearance of Hong Kong influenza in 1968. Colin and Theo determined the complete amino acid sequence of the A/duck/Ukraine/1/63 HA and showed that it indeed conformed to the H3 Hong Kong sub-type as predicted.
These sequence comparisons provided the first detailed, structural evidence to support the Laver-Webster theory of new influenza virus sub-types being generated by genetic re-assortment during mixed infections with human and avian influenza viruses. We now know that there are at least 15 HA and 9 NA subtypes in nature. The combinations H1N1, H2N2 and H3N2 account for the three major human pandemics seen since the influenza virus was first isolated in the 1930s. The emergence of Asian influenza in 1957 was associated with the exchange of three of the eight gene segments while in the generation of Hong Kong influenza in 1968 only the HA and PB1 genes were new, coming from an avian influenza virus; the other six gene segments were the same as those found in the pre-existing Asian flu variants. Bird influenza has the combination H5N1 and, while highly lethal in man, has not yet evolved to be readily transmissible from person to person. Swine influenza has the combination H1N1, similar to that seen in the 1918 Spanish influenza, the most serious recorded pandemic in history.
Immunochemical studies
The CSIRO program also included immunochemical analyses of HA fragments in collaboration with Dr David Jackson and Professor David White, Melbourne University. These studies clearly showed that the heavy chain, HA1 was the major immunogenic region of HA.
In addition, they showed that the isolated heavy chain HA1 contained the lectin-binding activity of influenza virus HA. In an unpublished haemagglutination-inhibition assay, Colin and Theo demonstrated that purified HA1 was able to bind to red blood cells and inhibit the binding of native virus in a competitive manner.
The structure of the influenza virus haemagglutinin – a landmark event
The CSIRO HA sequence data was also important for the interpretation of the electron density maps used in the determination of the 3D structure of the haemagglutinin protein by Don Wiley, Ian Wilson (Harvard University, Boston) and John (later Sir John) Skehel (National Institute for Medical Research, Mill Hill, London). This structure was published in Nature in 1981 and revealed that the accumulated sequence changes in the H3 haemagglutinin clustered into five main antigenic regions on the HA surface.
A further aid to the crystallography was Ward and Dopheide’s determination of the disulphide bond arrangements in HA; the location and nature of the seven attached oligosaccharide sub-units of this glycoprotein and the precise location of the C-terminal bromelain cleavage site of the solubilised influenza virus hemagglutinin trimers used for the crystallisation experiments.
Structural predictions of the presence of a coiled-coil in the HA trimers
In a short paper published in 1980 (before the 3D structure had been determined), Ward and Dopheide predicted (using amino acid sequence analyses), the existence of an extensive α-helical region in the HA light chain (HA2). They also detected the classical heptad repeat that is characteristic of coiled coils in proteins. The possibility of coiled coils in proteins was first proposed by Francis Crick in 1952 soon after the structure of the α-helix was suggested in 1951 by Linus Pauling and coworkers.
It was known that HA existed as non-covalently linked trimers on the virus surface, and Ward and Dopheide postulated that a folded-over, distorted coiled-coil might be responsible for this assembly. From published EM images they knew the long α-helical region of HA2 had to be folded over. They also concluded that there was potential for an even more extensive coiled-coil covering the stretch of 100 amino acids from residue 34 to 133.
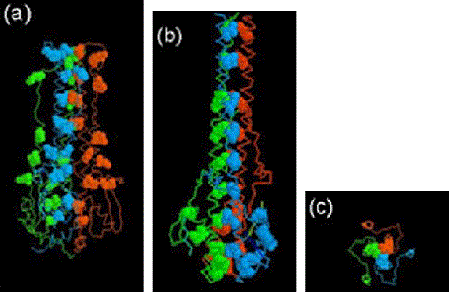
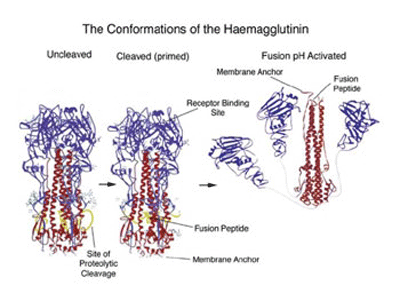
When the 3D structure of HA was finally solved by Wiley, Wilson and Skehel in 1981, a distorted, folded over, coiled-coil was indeed found to be holding the HA monomers together. One of the referees of the predictions paper felt that it should be discussed in ‘News and Views’ in Nature. It was, 15 years later in the 1 September 1994 issue, where the 3D structure of the fusogenic form of the influenza haemagglutinin was reported by Bullough, Hughson, Skehel & Wiley. This paper revealed how in the fusogenic form, the N-terminal portion of the haemagglutinin light chain (HA2) is released into an extended form with a more extensive coiled-coil that includes the distorted region (residues 57-77) predicted by Ward and Dopheide in 1980. This structural transition contributes to the process of fusing the viral and cellular membranes enabling the release of viral RNA into the infected cell.
The structural changes in the influenza virus haemagglutin trimer, from biosynthesis to infection are illustrated in the figure below. The alpha helical/coiled-coil regions of the three HA2 chains are shown in red. In the acidic conditions of the endosome after internalisation the three HA1 heads (blue) fall away allowing the folded over, distorted helical HA2 cores to swing up, generating the extended coiled-coil with the fusion peptide now at the top of the picture.
Invitations to write reviews on influenza coat proteins
In recognition of his contributions to the field Colin Ward was invited to write the first review (74 pages) on the 3D structure and sequence changes of the influenza virus haemagglutinin – published in 1981, Ward CW, 1981, Curr. Topics Microbiol. Immunol. 94/95, 1-74, and the corresponding review (79 pages) on influenza virus neuraminidase structure and variation – published in 1985, Colman PM, Ward CW, 1985, Curr. Topics Microbiol. Immunol. 114, 177-255.
Sources
- Walker R, 2004, Biographical entry: Fazekas de St Groth, Stephen Nicholas Emery Egon (Encyclopedia of Australian Science)
- Ward CW, 2010, Personal communication.