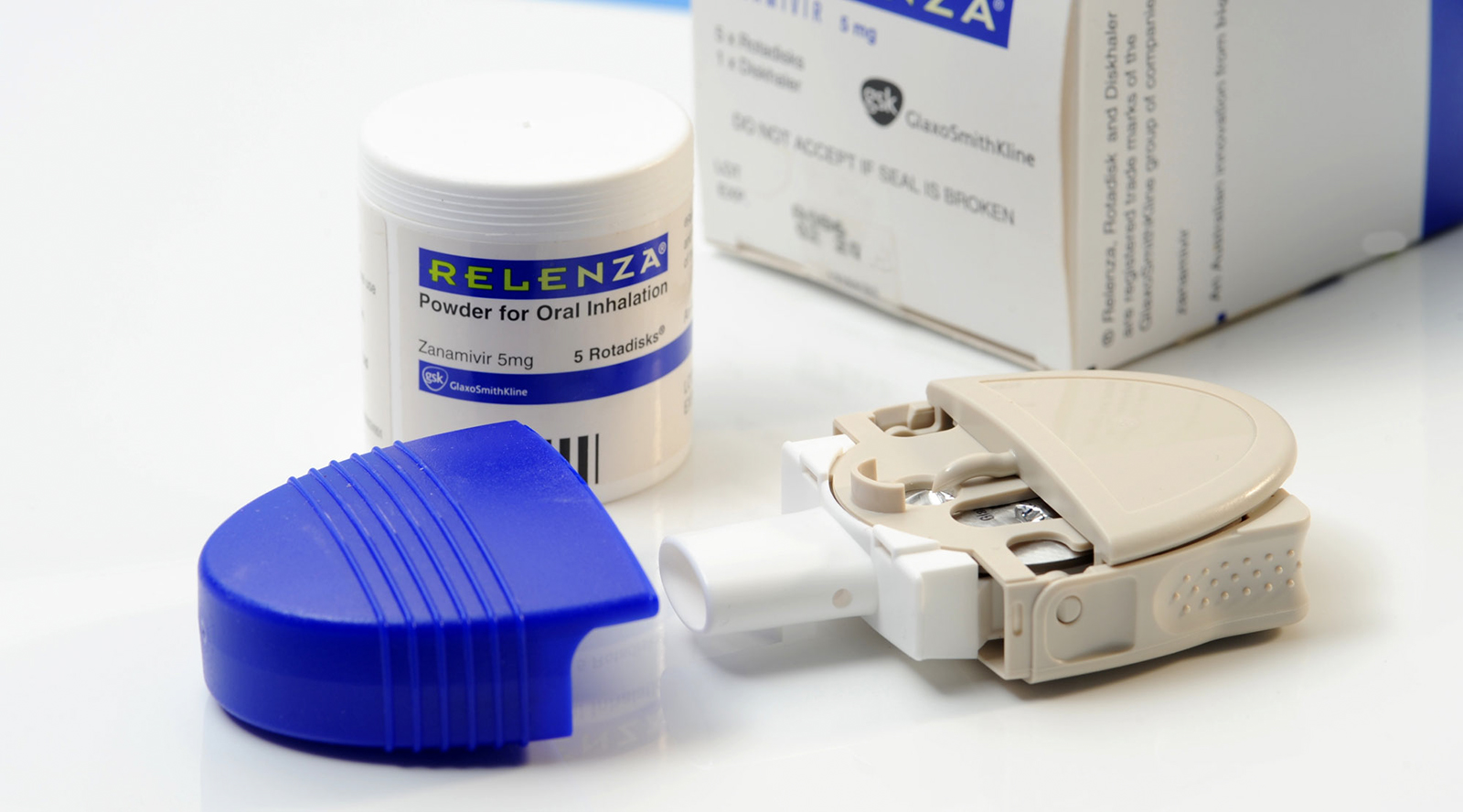
Relenza®
The determination of the three dimensional structure of the neuraminidase protein by Peter Colman and his colleagues at CSIRO in the 1980s, and its use in the development of the anti-influenza drug zanamavir (Relenza®) is a landmark medical discovery. Relenza® and the closely related oseltamivir (Tamiflu®) subsequently developed by the US-based company Gilead Sciences in collaboration with Roche, were both designed on the CSIRO neuraminidase structure and both drugs work against all strains of influenza virus including bird flu and swine flu.
These drugs, called neuraminidase inhibitors, represent a new class of anti-viral agent and have world-wide annual sales in excess of $3 billion. They target the enzyme, neuraminidase, which is found on the surface of all strains of the influenza virus. Neuraminidase plays an important role in releasing new virus particles from the surface of infected cells, enabling the virus to spread.
The research with collaborators at the Victorian College of Pharmacy, and the Australian National University is one of the first examples of structure-based drug design where the structure of the target protein is used to guide the chemistry.
In 1993 Time Australia ran the story ‘Through with Flu’ with Peter Colman and Mark von Itzstein (Victorian College of Pharmacy) featured on the cover. In 1996 Colman, von Itzstein and Graeme Laver (ANU) shared the Australia Prize for this achievement.

The influenza virus
Influenza, the flu, affects up to 500 million people each year. The virus has a significant economic impact in terms of days off work and lost productivity, and can also lead to death in the elderly or infirm. In a typical Australian winter, around 1,500 deaths are attributed to the flu.
The flu virus differs from most viral infections in being able to cause repeated infections in the same patient during their lifetime. In contrast a bout of measles, mumps or polio induces life-long immunity to the virus responsible.
By the early 1970s there was considerable speculation about the way in which the flu virus escaped immune responses and continued to cause repeated infections year after year. Attention turned to the surface of the flu virus, or more specifically, to the two proteins on its surface. Haemagglutinin, the major protein, is responsible for cell attachment and infection, while neuraminidase, the minor protein, aids the release of new virus particles from the surface of infected cells, enabling the virus infection to spread.
Early work
Initially most research focused on haemagglutinin as it was known to be the main target of protective immune reactions. CSIRO scientists Colin Ward and Theo Dopheide (CSIRO Protein Chemistry) and Gerry Both and Merilyn Sleigh (CSIRO Molecular Biology) provided much of the comparative amino acid and gene sequence data that explained the molecular basis of antigenic variation in the haemagglutinin. Their sequence data was also important for the determination of the 3D structure of the haemagglutinin protein by Don Wiley and Ian Wilson at Harvard University, a major achievement published in Nature in 1981.
In contrast neuraminidase was largely neglected, although it became a major interest for Peter Colman when he was appointed to establish X-ray crystallography at CSIRO’s Protein Chemistry Division in Parkville in 1978. At this time, there was a major influenza virus program in place at the Division being run by Colin Ward. Peter was joined on the neuraminidase crystallography project by Jose Varghese in 1981.

Seeing neuraminidase in 3D
A major milestone in the project was when the experiments with crystals of heavy metal derivatives of neuraminidase showed that the problem was solvable. Peter Colman describes the moment as follows:
The work was computationally intensive and weekends were good times to compute. Traffic on the line to the central machine in Canberra was low and competition for cpu time was minimal. One Saturday afternoon in 1982, after beating my way through the football traffic jam around Princes Park, I watched results come clanking back over the old line printer. I saw what I wanted and realised my goal was achievable, not the result itself, but apparently the avenue to it. At exactly the moment as the Line Printer fell silent, and my then five year old son played quietly on the floor with a box of disused punch cards, a cheer went up from 30 000 people at Princes Park ‘ and I wondered was there anywhere else in the world that the inexorable advance of science would be so welcomed.
An important component of the crystallography work to resolve the structure of neuraminidase was the availability of amino acid sequence data to aid the interpretation of the electron density maps. Over a two year period Colin Ward, Ahmed Azad and Tom Elleman cloned and sequenced the neuraminidase gene of one of the earliest 1957 Asian influenza variants. They also determined the amino acid sequence of the pronase-released heads of the neuraminidase from one of the last (1967) Asian flu epidemic strains, using protein chemical techniques.
Comparison of these two sequences, from viruses isolated 10 years apart, indicated the nature of the changes occurring in that period and revealed that the rate of change in the neuraminidase protein was the same as that seen in the major coat protein, the haemagglutinin.
One of the defining moments of the project occurred on the evening of Friday 17 August 1982 when Peter Colman and Jose Varghese finally saw how to connect up the various fragments of electron density into a polypeptide consistent with the sequence data provided by Ward, Elleman and Azad. The 3D structures of the two Asian flu viruses were published on 11 May 1983 in back-to-back papers in Nature.

A template for drug design
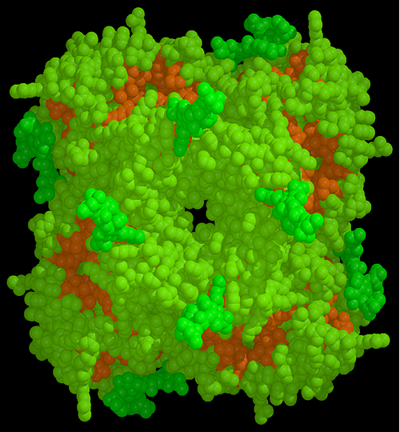
As shown in the figure below the structure revealed that the active site of the neuraminidase enzyme was totally conserved in all strains of human and animal influenza viruses. This led Peter Colman to suggest that, if the structure of this site could be refined to greater resolution, it could be used as a template to design an anti-influenza inhibitor that should be effective against all strains and subtypes of influenza neuraminidase.
Influenza virus replication involves a process where progeny viruses bud out of the plasma membrane of infected cells. Detachment of the newly formed virus from these buds requires the action of the viral neuraminidase to cleave off the terminal sialic acid residues from the membrane glycoproteins to which the virus is attached by its other viral surface glycoprotein the haemagglutinin. This removes the means for local attachment at the bud resulting in dispersal of the virus to infect other parts of the respiratory tract. Blocking viral neuraminidase would prevent this process and result in a subclinical infection which is terminated by the animals immune system before the infection has developed to give the severe symptoms of influenza.
Refining the structure was a major undertaking which began in 1983 and was completed in 1987.
Drug development
Peter Colman discussed the possibility of using the 3D structure for the development of a neuraminidase inhibitor with Glaxo in London in 1983. However, with previous attempts at developing this type of drug having failed, the pharmaceutical company was not interested. Instead, the development and testing of new neuraminidase inhibitors against influenza virus was undertaken in Melbourne, supported by Biota Holdings Limited, which was floated on the Australian Stock Exchange at the end of 1985. The drug development program involved a collaboration between Peter Colman’s group at Parkville and Peter Andrews’ group at the Department of Medicinal Chemistry, Victorian College of Pharmacy (now part of Monash University).
After initial unsuccessful attempts to design inhibitors using SAR techniques, the breakthrough came when the crystallographic structures of complexes of the influenza virus neuraminidase with sialic acid (N-acetyl neuraminic acid, NANA) and 2-deoxy-2,3-didehydro-N-acetylneuraminic acid (DANA) were solved by CSIRO in 1992. It was known, as far back as 1974, that DANA, a sialic acid analogue, was an inhibitor of neuraminidase. Sialic acid (N-acetyl neuraminic acid, NANA), the substrate of neuraminidase, is itself a mild inhibitor of the enzyme, but the dehydrated derivative DANA, a transition-state analogue, is a better inhibitor. Following a suggestion from Peter Colman that, GlaxoSmithKline (GSK) test DANA as an anti-viral in their Ferret animal model, it was found that DANA did indeed show some inhibition of viral spread. This led to a more formal relationship between Biota and GSK.
The landmark paper on the design and evaluation of the inhibitor zanamavir was published in Nature in 1993. The design strategy relied on the availability of the X-ray crystal structure of the complex of influenza neuraminidase and DANA. Computational chemistry techniques were used to probe the active site of the enzyme, in an attempt to design derivatives of DANA that would bind tightly to the aminoacid residues of the catalytic site, and so would be potent and specific inhibitors of the enzyme. The software GRID from Molecular Discovery was used to determine energetically favourable interactions between various functional groups and residues in the catalytic site canyon. This showed there was a negatively charged zone in the neuraminidase active site that aligned with the C4 hydroxyl group of DANA. This hydroxyl was therefore replaced with a positively charged amino group; the 4-amino DANA was 100 times better an inhibitor than DANA, owing to the formation of a salt bridge with a conserved glutamic acid (Glu119) in the active site. It was also noticed that Glu119 was at the bottom of a conserved pocket in the active site just big enough to accommodate a more basic functional positively charged group, such as a guanidino group, which was also larger than the amino group. Zanamivir, a transition-state analogue inhibitor of neuraminidase, was the result.
Zanamavir, the 4-guanadino analogue of DANA, was synthesised by Wen-Yang Wu, in Mark Von Itzstein’s laboratory at the Victorian College of Pharmacy and was found to be a potent inhibitor of influenza virus replication in cells and in the ferret animal model.

Relenza® hits the shelves
Following the success of this study and indications that Relenza® was a poor inhibitor of endogenous human and animal neauraminidases, the compound was then successfully trialled in humans. Zanamavir entered into Phase I clinical trials in 1993, Phase II clinical trials in 1994 with the Phase III clinical trials completed in 1997. The application for US marketing of zanamavir under the trade name Relenza® was filed in 1998 and FDA approval obtained in the same year. The drug was launched world-wide in 1999 and has been approved in over 50 countries for the treatment of influenza. It is also widely approved for use as a preventative treatment against influenza.
CSIRO’s ongoing involvement
CSIRO’s involvement with Relenza® continues through Dr Jenny McKimm-Breschkin, who contributes to the world-wide surveillance program on the emergence of drug resistant influenza variants.
Sources
- Rivett DE, Ward CW, Belkin LM, Ramshaw JAM, Wilshire JFK, 1996, ‘The Lennox Legacy ‘ A History of the CSIRO Laboratory at 343 Royal Parade Parkville’, CSIRO Publishing, Melbourne, Australia, pp370.
- Colman PM, Ward CW, 2008, Personal communication.
- Varghese JN, 2010, Personal communication.